
Congenital Adrenal Hyperplasia (CAH)
Prognosis and Outlook
The long-term outlook (prognosis) for people with congenital adrenal hyperplasia (CAH) is usually good. With lifelong treatment, affected people typically have good health and normal life spans. However, problems with psychological adjustment are common, particularly in people with genital abnormalities. If congenital adrenal hyperplasia (CAH) is treated soon after birth, children can have healthy growth and development. This is why newborn screening is so important.
Early puberty is a common sign of CAH in childhood. Children who receive treatment may still have slightly early puberty. Adults with CAH may have shorter stature than other members of their family, even with treatment.
Babies who do not receive treatment for salt-wasting CAH are at risk for coma or death.
Incidence
Global Incidence
The estimated prevalence is 1/10,000 and annual incidence ranges from 1/5,000 to 1/15,000 (ref: Orpha). In the United States, about one in every 15,000 babies is born with congenital adrenal hyperplasia (CAH). The condition may be more or less common in certain ethnic groups and geographic regions. (Baby’s First Test)
Congenital adrenal hyperplasia caused by 21-hydroxylase deficiency is found in all populations. 11-beta-hydroxylase deficiency is more common in persons of Moroccan or Iranian-Jewish descent. (Medscape)
Factors that increase the risk of having congenital adrenal hyperplasia include people in certain ethnic groups, particularly Ashkenazi Jews, but also Hispanics, Italians, Yugoslavians and Yupik Inuits. (Mayoclinic)
Local Incidence
A study was conducted in 2010 by Military Hospital Rwaalpindi, and they reported it is a recessively inherited disorder and has an average incidence of 1:5000; the most common of these is 21-hydroxylase deficiency that accounts for 95% of involved cases.
“There is a prompt necessity to setup national and regional neonatal screening programswhile including CYP21A2 genotyping in the diagnosticcomponent to address this serious health issue” (paper: Ethnic disparity in 21-hydroxylase gene mutations identified in Pakistani congenital adrenal hyperplasia patients)
“Children with CAH should be diagnosed early as a rational and judicious choice of sex assignment is a critical aspect of treatment. The high rate of consanguinity emphasized the need to establish the true incidence of the defect in Pakistani population” (JPMA 54:509;2004).
Parental consanguinity was found in 33 (52.3%) cases in study published in October 2004. Genetic studies indicate that boys and girls are at equal risk for CAH. However, the prevalence of CAH is higher among girls than boys with this disease. Males with the non-classical form remain undiagnosed (Bhanji, R et al 2004).
What is CAH?
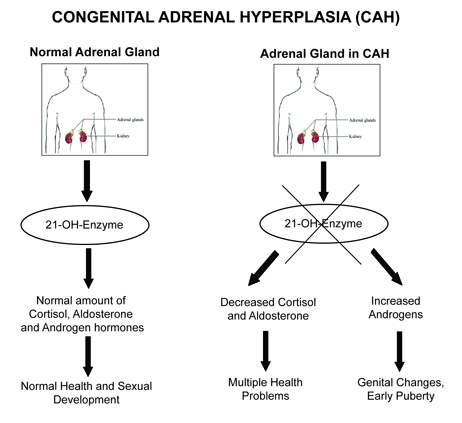
Congenital adrenal hyperplasia (CAH) is a group of inherited genetic disorders that affect the adrenal glands, a pair of walnut-sized organs above your kidneys. A person with CAH lacks one of the enzymes the adrenal glands use to produce hormones that help regulate metabolism, the immune system, blood pressure and other essential functions.
21-hydroxylase deficiency is responsible for 95% of all cases of CAH and is broken down further into two subcategories: classical CAH, which can be sub-divided into the salt-losing form or the simple-virilizing form, and non-classical CAH.
Others Forms
Subdivisions of Congenital Adrenal Hyperplasia include:
- 11-Beta hydroxylase deficiency : patients are protected from the symptoms associated with adrenal crisis, although they are subject to others such as hypertension due to salt retention and ambiguous genitalia in females.
- 17a-hydroxylase deficiency : results in ambiguous external genitalia in males and lack of pubertal development or menstrual cycles (amenorrhea) in females.
- 3-Beta-hydroxysteroid dehydrogenase deficiency : leads to ambiguous genitalia in males and females. In both genders it can lead to salt-wasting.
- congenital lipoid adrenal hyperplasia : may cause early death due to adrenal crisis. Males have ambiguous genitalia. Both males and females, if they survive, would likely be infertile.
- PORD (P450 oxidoreductase deficiency) presents with signs and symptoms that may resemble 21-hydroxylase deficiency, 17-hydroxylase deficiency, or a combination of the two enzyme deficiencies. Some cases have been associated with a skeletal disorder known as Antley-Bixler syndrome.
Types
There are two types of CAH—classic, which can be life threatening, and nonclassic, a milder form of the disorder.
Classic CAH
Classic CAH, usually first found in infancy or early childhood, is the most severe type of CAH. In one form of classic CAH, called “salt-wasting” (meaning the body has trouble keeping the right amount of salt in the blood), the adrenal glands do not make enough cortisol and aldosterone. If not found and treated, classic CAH can cause shock, coma, and death. In another form of classic CAH, called “non-salt wasting,” the enzyme shortage is less severe. The adrenal glands make just enough aldosterone but not enough cortisol.
Signs and symptoms
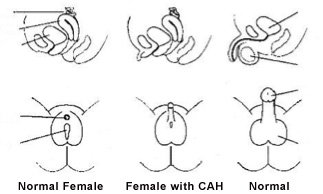
In many cases, female infants are diagnosed at birth because they have atypical or ambiguous genitalia (external sex organs that resemble male genitals). However, they still have normal internal female organs (ovaries and uterus).
A male infant with classic CAH usually appears normal at birth, although he may have an enlarged penis. If a boy is not diagnosed in infancy, he may grow rapidly and show signs of early puberty.
Any infant, boy or girl, who is not diagnosed and treated appropriately at birth, may have vomiting, weight loss, dehydration, shock, and even death.
Non-classic CAH
Unlike classic CAH, non-classic CAH is mild and not life threatening. Signs and symptoms might not appear until childhood or adulthood.
Signs and symptoms
Signs and symptoms in both males and females include:
- Early development of armpit and pubic hair
- Rapid growth during childhood
- Early or severe acne
- Infertility or decreased fertility
- Adolescent girls and adult women also may have:
- Masculine characteristics such as facial hair, acne and/or a deep voice
- Infrequent or absent menstrual periods
Testing and follow-up
Newborn Screening:
If your baby’s newborn screening result for congenital adrenal hyperplasia (CAH) was out of the normal range, your baby’s doctor or the state screening program will contact you to arrange for your baby to have additional testing. It is important to remember that an out-of-range screening result does not necessarily mean that child has the condition.
Follow up testing:
Follow-up testing will involve checking your baby’s urine and blood samples for signs of CAH. Children with CAH have high levels of a chemical called 17-OHP in their blood because they are unable to turn this chemical into cortisol, a hormone produced in the adrenal glands. High amounts of 17-OHP in the blood might indicate that your baby has CAH. Further testing, which may include genetic testing, can determine which form of CAH your child has.
How is CAH diagnosed?
After Birth or Newborn Screening Testing
Some forms of CAH are diagnosed at birth. However, milder forms might not be diagnosed until childhood or adulthood. In the United States and many other countries, all newborns undergo a blood test for the most common form of classic CAH. Diagnosis in infancy or later also may include. Further diagnosis includes:
- Urine tests
- Genetic tests
- A physical examination
- A family history
- Physical exam.The doctor examines your child and evaluates symptoms. If, based on these findings, the doctor suspects CAH, the next step is to confirm the diagnosis with blood and urine tests.
- Blood and urine tests.Tests used to diagnose CAH measure levels of hormones produced by the adrenal glands. A diagnosis can be made when there are abnormal levels of these hormones.
- Gene testing.In older children and young adults, genetic testing may be needed to diagnose CAH.
- Testing to determine a child’s sex.In female infants who have severe ambiguous genitalia, tests can be done to analyze chromosomes to identify genetic sex. Also, pelvic ultrasound can be used to identify the presence of female reproductive structures such as the uterus and ovaries.
Diagnosis availability
Genetic test services are available in the Islamabad city, like:
- Congenital adrenal hyperplasia due to 17-alpha-hydroxylase deficiency (deletion/duplication analysis on CYP17A1 gene)
- Congenital adrenal hyperplasia due to 17-alpha-hydroxylase deficiency (sequence analysis of CYP17A1 gene)
The information on test will be conveyed on inquiry.
Treatment
Congenital adrenal hyperplasia can be life threatening if left untreated. Quality healthcare and treatment services provided at the Internal Medicine Service Line at the Aga Khan University Hospital, Karachi, can help ensure that your child leads a normal healthy life.
Your treatment options will include:
- Hormone replacement therapy to replace the levels of deficient hormones such as, hydrocortisone or dexamethasone to replace cortisol, and fludrocortisone to replace aldosterone. Your child may have to take the medications orally every day. They may also have to take a higher dosage in cases of illness or severe stress, including surgery.
- Regular monitoring by the doctor and blood tests to ensure that the drugs are being given in the correct dosage and not causing any side effects.
- Constant observation to look for signs of infections or side-effects of the medications, which are usually rare.
- Surgery for infant girls with ambiguous outer genitals, to correct the appearance and functions of the genitals.
Your child’s doctor may prescribe replacement hormone medication taken on a daily basis to restore normal levels of deficient hormones. Additional medications or higher doses may be needed during periods of illness or significant stress such as surgery.
Prenatal Treatment
It is for CAH should be regarded as experimental.
- Glucocorticoid therapy should be carefully titrated to avoid Cushing syndrome.
- Mineralocorticoid replacement is encouraged. In infants, mineralocorticoid replacement and sodium supplementation are encouraged.
- Use of agents to delay puberty and promote growth are experimental.
- Psychiatric support should be encouraged for patients with adjustment problems.
- Medication should be used judiciously during pregnancy and in symptomatic patients with nonclassical CAH.
Non-classical CAH on the other hand, is not life-threatening and relatively mild. People who have no obvious symptoms of non-classical CAH do not require surgery or medical treatment. If a patient with non-classical CAH begins to enter puberty too early, has early maturation of bones, or is a female with excess facial or body hair or other masculine features, glucocorticoid treatment is recommended. Fertility problems can also be corrected with glucocorticoids and/or fertility drugs. Women who do not wish to conceive may also be prescribed oral contraceptives. Unlike severe forms of CAH, non-classical CAH patients are free to taper and stop treatment when symptoms go away.
Diet
- Patients with congenital adrenal hyperplasia should be on an unrestricted diet.
- Patients should have ample access to salt because salt wasting is common in some forms of the disease.
- Infants who have salt wasting generally benefit from supplementation with NaCl (2-4 g/d) added to their formula.
- Caloric intake may need to be monitored and restricted if excess weight gain occurs because glucocorticoids stimulate appetite.
Medications
Patients with dehydration, hyponatremia, or hyperkalemia and a possible salt-wasting form of adrenal hyperplasia should receive an intravenous (IV) bolus of isotonic sodium chloride solution (20 mL/kg or 450 mL/m 2) over the first hour, as needed, to restore their intravascular volume and blood pressure.
- This dosage may be repeated if the blood pressure remains low.
- Dextrose must be administered if the patient is hypoglycemic and must be included in the rehydration fluid after the bolus dose to prevent hypoglycemia.
- After samples are obtained to measure electrolyte, blood sugar, cortisol, aldosterone, and 17-hydroxyprogesterone concentrations, the patient should be treated with glucocorticoids based on suspected adrenal insufficiency. Treatment should not be withheld while confirmatory results are awaited because it may be life preserving.
After the patient’s condition is stabilized, treat all patients who have adrenal hyperplasia with long-term glucocorticoid or aldosterone replacement (or both), depending on which enzyme is involved and on whether cortisol and/or aldosterone synthesis is affected.
Another approach currently under investigation is the combined use of glucocorticoid (to suppress ACTH and adrenal androgen production), mineralocorticoid (to reduce angiotensin II concentrations), aromatase inhibitor (to slow skeletal maturation), and flutamide (an androgen blocker to reduce virilization).
Some patients develop precocious puberty, which further compromises adult height. Suppression of puberty with long-acting gonadotropin-releasing hormone (GnRH) agonists while simultaneously stimulating growth with growth hormone may partially improve the patient’s height.
The Endocrine Society’s 2010 clinical practice guidelines note the following:
- Prenatal treatment for CAH should be regarded as experimental.
- Glucocorticoid therapy should be carefully titrated to avoid Cushing syndrome.
- Mineralocorticoid replacement is encouraged. In infants, mineralocorticoid replacement and sodium supplementation are encouraged.
- Use of agents to delay puberty and promote growth are experimental.
- Psychiatric support should be encouraged for patients with adjustment problems.
- Medication should be used judiciously during pregnancy and in symptomatic patients with nonclassical CAH.
Surgery
Infants with ambiguous genitalia require surgical evaluation and, if needed, plan for corrective surgery. The traditional approach to the female patient with ambiguous genitalia due to adrenal hyperplasia is clitoral recession early in life followed by vaginoplasty after puberty. Some female infants with adrenal hyperplasia have only mild virilization and may not require corrective surgery if they receive adequate medical therapy to prevent further virilization.
A retrospective study by Dangle et al on feminizing reconstructive surgery in 26 toddlers with congenital adrenal hyperplasia reported that the procedures are typically well tolerated and successful. Although complications were found in seven patients (27%), including dysuria, stitch dehiscence, wound separation, and urinary tract infection, revision surgery was needed in only two individuals (7.7%). The patients were followed up for an average of about 6 years.
Bilateral adrenalectomies have been suggested in the management of virilizing forms of adrenal hyperplasia in order to prevent further virilization and advancement of skeletal maturation. This approach is experimental and should be considered only in the context of a controlled study.
The Endocrine Society’s 2010 clinical practice guidelines note the following:
- Adrenalectomy should be avoided.
- While surgical reconstruction may not be necessary during the newborn period in mildly virilized girls, it may be appropriate in severely virilized girls. It should be a single stage genital repair, performed by experienced surgeons.
References
- https://www.mayoclinic.org/
- https://rarediseases.org/
- http://www.babysfirsttest.org/
- Yang M, White PC. Risk factors for hospitalization of children with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2017 Feb 13.
- https://rarediseases.org/
- Journal Of Pakistan Medical Association
- Pakistan Armed Forces Medical Journal
Leave a Reply